
The Importance of Bioinformatics in Molecular Biology Research (Part II)
Part II: Network Analysis & Molecular Docking, Simulation
Nur Alyaa Afifah Md Shahri, Nur Nadia Razali & Saiful Effendi Syafruddin
UKM Medical Molecular Biology Institute, Universiti Kebangsaan Malaysia
Date: 5 April 2021
Bioinformatics enables researchers to explore and make novel and significant discoveries in the field of biological and medical sciences. Hence it is worth learning and embracing the relevant bioinformatics skills and applications to consolidate our research and data analysis. In this part II, will introduce and focus on discussing the Network Analysis & Molecular Docking and Molecular Dynamics Simulation.
Network Analysis
Network analysis is a bioinformatic technique that allows researchers to elucidate the putative interactions, which is also known as interactome, between the subcellular of interest. The biological network facilitates the understanding of cellular components and subprocesses as interacting systems (1,2). There are many molecular mechanisms inside the cells that involve intricate protein-protein interactions, which include replication, transcription and translation regulations, intracellular signalling cascades, metabolic pathways and so on. In this article, we will be focusing on analysing protein-protein interaction networks (PPIN) by computational approach.
There are a number of network analysis tools that can be used to integrate, visualize and analyze the interaction between the subcellular components (3). For example, Cytoscape is widely used in network analysis because it provides various apps for specific functionality to the core distribution of Cytoscape. It makes this tool adaptable to multiple types of analysis (2). There are specific apps available for PPIN in Cytoscape, such as MCODE, clusterMaker2, JActiveModules, and CentiScaPe. However, Cytoscape has their limitation when it comes to large-scale networks (hundreds of thousands of nodes and edges). Thus, there are two options to solve this issue which are using the non-programmatic tool (Gephi) or programmatic solution such as igraph (for R, Python, and C), NetworkX (for Python), and graph-tool (2).
Table 1 below shows the list of several available databases for network analysis for PPIN and their classification based on the data-acquisition policies (2,4). Moreover, the general workflow to perform PPIN analysis is exemplified in the figure 1 below (2, 3, 5, 6, 7).
Table 1: Available database for PPIN.
| Classification | Name of database | Data type | Website Link |
| Primary Database | IntAct Molecular Interaction Database (IntAct) | DNA, Protein, RNA | https://www.ebi.ac.uk/intact/ |
| Molecular Interaction Database (MINT) | Protein | https://mint.bio.uniroma2.it/ | |
| Biological General Repository for Interaction Datasets (BioGRID) | DNA, Protein | https://thebiogrid.org/ | |
| Secondary Database / Metadatabase | Agile Protein Interactomes DataServer (APID) | Protein | http://cicblade.dep.usal.es:8080/APID/init.action |
| Protein Interaction Network Analysis (PINA) | Protein | https://omics.bjcancer.org/pina/ | |
| Unified Human Interactome (UniHI) | Protein | http://www.unihi.org/ | |
| Prediction Database | Search Tool for the Retrieval of Interacting Gene (STRING) | Protein | https://string-db.org/ |
| Human protein-protein interaction prediction database (PIPs) | Protein | http://www.compbio.dundee.ac.uk/www-pips/ | |
| Interologous Interaction Database (I2D) | Protein | http://ophid.utoronto.ca/ophidv2.204/ |
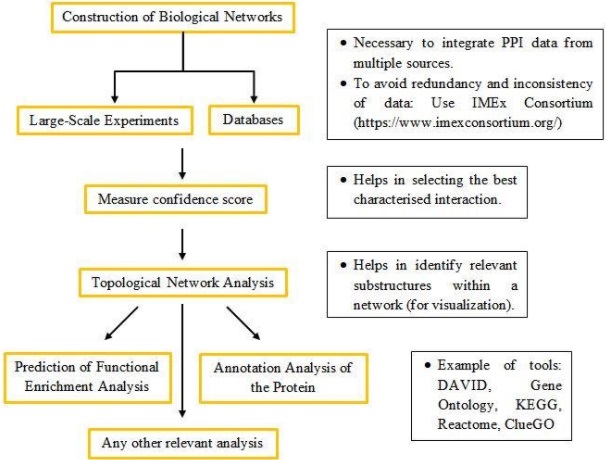
Figure 1: Common steps in performing PPIN analysis.
In conclusion, the network analysis is essential to extract meaningful information from the complexity of the networks (2), to predict the function and behavior of a new protein-based on its structure (8), to predict the biological processes of a protein with unknown function (1), to understand biological mechanisms and disease etiologies (3) as well as for the development of new drugs and discovery of disease pathways (9).
Molecular Docking and Dynamics
Subsequently, the interaction between molecules and their respective interactome obtained from the network analysis can be predicted and visualized using molecular docking and simulation algorithms. The understanding of docking at the molecular level is straightforward where two or more macromolecular structures such as enzyme, drug or protein fit together. Modelling the interaction between molecules is among the important aspects in the field of structural biology. It involves computational algorithms and approaches that predict the molecules’ mutual configuration position, and ability to form a complex. One important application of molecular docking tools is in drug development. This includes predicting the binding orientation, dynamic and efficiency of the developed small inhibitors to their target sites within the proteins of interest which force will fit with it, and how to screen it against time. The docking scene will illustrate the three-dimensional protein structures and the interaction with the targeted ligand, as shown in figure 2 (10).

Figure 2: General procedures for molecular docking
The interaction and activity between the molecules of interest can be assessed using conventional biochemical assays as well as using more high-end instrumentation, such as the liquid chromatography coupled with mass spectrometry. Therefore, in order to visualize and elucidate the mechanism of macromolecule-ligand bonding and interaction, in silico methodology is applied. The rationale to employ in silico research reduces wet-lab experiments’ uncertainty output, focusing on promising candidates containing the potential in target features (11). The molecular docking is needed to explain what is appealing behind the complexing of biology in the human body.
A protein like Cytochrome P450 protein, a giant protein in size, interacts with a diverse range of drugs and undergoes detoxification (12). Therefore, there is a step to understand the mechanism, known as molecular docking. What happens in molecular docking and dynamic? It is a routine task to improve the 3D structure from homology models (13). This calculation involves evaluating the stability and folding when it is in a solvent, mimicking the real physiological environments. Some scoring function measures will justify the simulation’s output in the dynamic simulation process to enhance the docking process’s qualitative analysis. There are many tools and softwares to do molecular dockings, such as Schrödinger (14), Desmond and Amber (15), and Gromac (16). These various simulation tools are available with high efficiency nowadays, which is fascinating to explore in several molecular research areas.
Massive improvements have been achieved in the sensitivity and efficiency of bioinformatics in the molecular biology approach. The new generation of bioinformatics technologies can emphasize high dimensional and dynamic details in the technique. From this point forward, a standardized procedure of bioinformatics with integrated-omics molecular analysis strategy using all the tools are available for various types of research. The devices can either be used from public websites or install the software as stand-alone tools on a computer.
References
- Altaf-Ul-Amin M, Afendi FM, Kiboi SK, Kanaya S. Systems biology in the context of big data and networks. Biomed Res Int. 2014;2014(3).
- Porras Millan P, Ochoa D, Z. Rogon M, Garcia Alonso L, Turei D. Network analysis of protein interaction data. EMBL-EBI Training. 2020.
- Zhang P, Itan Y. Biological network approaches and applications in rare disease studies. Genes (Basel). 2019;10(10):1–15.
- Taghizadeh M, Safari-Alighiarloo N, Tavirani MR. Protein-Protein Interaction Database: An Overall View on Interactome Organization the Nature of Protein-Protein Interaction Data. Int J Anal Pharm Biomed Sci. 2015;4(1):15–23.
- Bozhilova L V., Whitmore A V., Wray J, Reinert G, Deane CM. Measuring rank robustness in scored protein interaction networks. bioRxiv. 2018;1–14.
- Chagoyen M, Ranea JAG, Pazos F. Applications of molecular networks in biomedicine. Biol Methods Protoc. 2019;4(1):1–10.
- Koutrouli M, Karatzas E, Paez-Espino D, Pavlopoulos GA. A Guide to Conquer the Biological Network Era Using Graph Theory. Front Bioeng Biotechnol. 2020;8(January):1–26.
- Porras Millan P. Protein interactions and their importance | EMBL-EBI Training. 2020.
- Muzio G, O’Bray L, Borgwardt K. Biological network analysis with deep learning. Brief Bioinform. 2020;00(April):1–17.
- Tech JAB, Dar AM, Mir S. Analytical & Bioanalytical Techniques Molecular Docking : Approaches , Types , Applications and Basic Challenges. 2017;8(2):8–10.
- Cesar B, Hinckel B, Marlais T, Airs S, Bhattacharyya T, Imamura H, et al. Refining wet lab experiments with in silico searches : A rational quest for diagnostic peptides in visceral leishmaniasis. 2019;1–20.
- Ogu CC, Maxa JL. Drug Interactions Due to Cytochrome P450. 2017;8280:11–4.
- Shen J, Li W, Liu G, Tang Y, Jiang H. Computational Insights into the Mechanism of Ligand Unbinding and Selectivity of Estrogen Receptors. 2009;10436–44.
- Ligands S, Karelson M. Molecular Dynamics Simulations of the Interactions between Glial Cell Line-Derived Neurotrophic Factor Family Receptor GFR α 1 and Small-Molecule Ligands. 2018;
- Id AR, Mahrous GM. Docking studies and molecular dynamics simulations of the binding characteristics of waldiomycin and its methyl ester analog to Staphylococcus aureus histidine kinase. 2020;1–16. Available from: http://dx.doi.org/10.1371/journal.pone.0234215
- Ferreira RJ, Ferreira MU, Santos DJVA. Insights on P-Glycoprotein’ s E ffl ux Mechanism Obtained by Molecular Dynamics Simulations. 2012;