
Genomic Surveillance Of SARS-CoV 2: Comparison between Whole Genome Sequencing and Variant Genotyping
Khairun Ghafar1, Nor Azila Muhammad Azami1 & Rahman Jamal1
1UKM Medical Molecular Biology Institute
In June 2021, a national consortium for genomic surveillance of SARS-CoV-2 was established as Malaysia prepared for the endemic phase. UMBI and six other institutes from three different ministries (Ministry of Health, Ministry of Sciences and Technology, and Ministry of Higher Education) were the initial team members of this national consortium funded by the Strategic Research Fund by the Ministry of Science, Technology, and Innovation (MOSTI). The head of this program was Professor Datuk Dr. A Rahman A Jamal from UMBI. From January 2022, a new grant was approved under the Program Strategik Memperkasa Rakyat dan Ekonomi (PEMERKASA) again under the MOSTI. For the second phase, the administrator for the funding is the Malaysia Genome and Vaccine Institute (MGVI), and four more institutions joined the consortium. Genomic surveillance is an important strategy to analyses, monitor, and track the variants of interest and their prevalence by detecting the lineages of circulating SARS-CoV-2 virus in the community transmission either by whole genome sequencing (WGS) or variant genotyping. The genomic surveillance is performed on samples that fulfilled the specific criteria and deemed as the representative of the affected population.
SARS-CoV-2 virus is a b-coronavirus with an enveloped non-segmented positive sense RNA virus. Like any other organism, genes in the SARS-CoV-2 genome code for different parts of the virus. The genome of SARS-CoV-2 encodes for four structural proteins including spike (S), envelope (E), membrane (M) and nucleotide (N) and several non-structural proteins (Figure 1). The S gene, for example, codes for the spike protein of the virus which is vital for its spreading ability (Illumina, 2022). Mutations commonly occur during virus replication due to lack of proofreading. Majority of these mutations do not affect the viral fitness, virulence, and epidemiology. However, once a mutation affects the virus abilities, the virus will become more fit and infectious, and has a competitive advantage over other lineages, and subsequently able to displace the existing circulating lineage in the population (Yin, 2020). During the COVID-19 pandemic, lineages that have an impact on public health will be classified as “variant of interest” (VOI) or “variant of concern” (VOC).
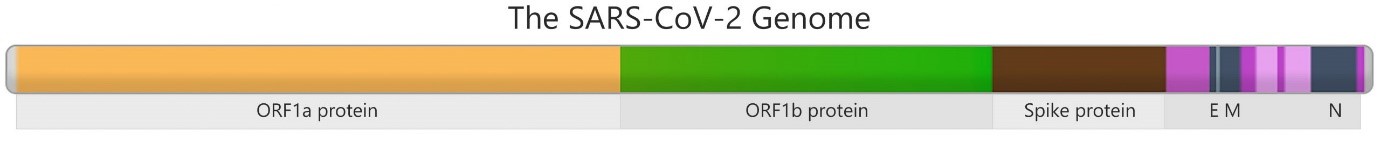 Figure 1: The SARS-CoV-2 Genome with S gene encoding for the spike protein
Figure 1: The SARS-CoV-2 Genome with S gene encoding for the spike protein
(Source: Illumina Website)
There are two methods widely used for the genomic surveillance: whole genome sequencing (WGS) and variant genotyping. WGS is a method to sequence and analyse an entire genome of an organism (Illumina, 2022). WGS method can be used to identify inherited disorders and track any mutation in human genome that cause cancer and non-communicable diseases, to find causative variants of pathogens in agriculturally important livestock, and to detect the presence of emerging virus such as the SARS-CoV-2 virus (Illumina, 2022). Results from the WGS can be used to detect single nucleotide variants, mutations by insertions or deletions and any small or large structural changes in the nucleotide sequence that might be missed with targeted approaches (Illumina, 2022). In recent years, the cost of WGS has reduced significantly and becoming affordable due to the advancement in sequencing technologies. The production of big data from WGS will require expertise in bioinformatics for analysis. In UMBI, the SARS-CoV-2 genome surveillance was performed using the Illumina Miseq and the Nanopore GridIon platform.
In the WGS method, the extracted RNA goes through a series of steps such as quality control check using quantitative Polymerase Chain Reaction, library preparation and sequencing. In the quality control steps, these samples need to pass certain criteria that was set by the kit. For the library preparation, the COVIDSeq kit or Nanopore Midnight library kit were used in UMBI. For the COVIDSeq library kit, the RNA samples used must have Ct value less than 27 while the Nanopore Midnight Protocol Library kit required RNA samples with a Ct value less than 30. After the quality control, the RNA samples will undergo the library preparation steps. The library preparation steps include Polymerase Chain Reaction (PCR), fragmentation and tagmentation (Illumina, 2022). After the library preparation, the sequencing will be performed using either Illumina Miseq or Nanopore GridIon to generate the FASTQ file. The generated FASTQ will be analysed using the Illumina DRAGEN COVID Pipeline Software for the Miseq platform or Epi2Me software for the Nanopore GridIon platform. The SARS-CoV-2 lineages will be assigned using the open-source bioinformatics tool such as Nextclade by Nextstrain or Phylogenetic Assignment of Named Global Outbreak Lineages by COVID-19 Genomics UK Consortium (PANGOLIN) (Figure 2). Although some sequencing platform comes with their analysis platform like Nanopore, it is always recommended to cross-check the results using other platforms as well as the database are constantly updated. Depending on the library kit and sequencing platform, the WGS method may take up from one to three days.
Figure 2: The interface of Nextclade tool (https://clades.nextstrain.org/) and Phylogenetic Assignment of Named Global Outbreak Lineages tool (https://pangolin.cog-uk.io/) used for lineage determination.
In contrast, the variant genotyping method is used to detect small genetic differences such as single-nucleotide polymorphism at a specific position within the genome that can lead to major changes in phenotype (Kwok & Chen, 2003). Based on the genotyping analysis, genes that might be associated with the SARS-CoV-2 virality and transmissibility such as the S proteins, RNA polymerase, RNA primase, and nucleoprotein genes were revealed to have the most frequent mutations (Yin, 2020). In the case of Covid-19 Genome Surveillance, variant genotyping will detect the specific multiple mutations that acted as the predominated mutation type by comparing the genome sequences to a reference sequence; for example, the N501Y and K417N mutations are common mutation for VOC Omicron (Yin, 2020). Unlike WGS that requires high-end sequencing equipment, variant genotyping method can be performed using real-time PCR method that requires more basic laboratory equipment such as the thermocycler machine. Comparing to the WGS method, variant genotyping method protocol is easier, cheaper, and faster with a higher throughput (Kwok & Chen, 2003). More importantly, the bioinformatic analyses are much less complex than WGS because the end result can be easily interpreted without the need of bioinformatics tools or pipeline (Figure 3). However, given the simplicity of genotyping, the results gained from this method must be validated using sequencing to ensure accuracy.
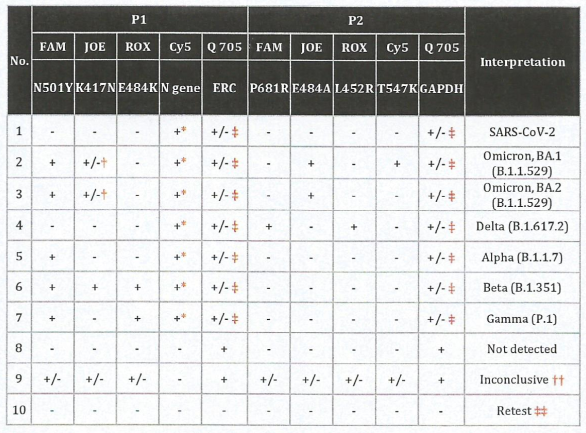
Figure 3: An example of interpretation table to determine variants of SARS-CoV-2 via genotyping.
In conclusion, in the genomic surveillance settings, both WGS and variant genotyping methods are suitable depending on the surveillance requirements. However, to set up a laboratory for genomic surveillance purpose, one must be considering various factors and resources such as sample size, cost, laboratory equipment, human resources, and time.
References
- Illumina. (2022). Introduction to Genotyping. Retrieved from Genotyping methods and solutions: https://sapac.illumina.com/techniques/popular-applications/genotyping.html
- Illumina. (2022). What is Whole-Genome Sequencing? Retrieved from A high-resolution view of the entire genome: https://sapac.illumina.com/techniques/sequencing/dna-sequencing/whole-genome-sequencing.html
- Kim, H. J., Kim, S. Y., Kwon, G. C., & Choi, Q. (2022). Detecting spread of SARS-CoV-2 variants using PowerChek SARS-CoV-2 S-gene mutation detection kit. Journal of clinical laboratory analysis, e24567. Advance online publication. https://doi.org/10.1002/jcla.24567
- Kwok, P. Y., & Chen, X. (2003). Detection of single nucleotide polymorphisms. Curr Issues Mol Biol, 5:43–60.
- Thermo Fisher Scientific. (2022). What is Genotyping? Retrieved from Thermo Fisher Scientific: https://www.thermofisher.com/my/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/genotyping-analysis-real-time-pcr-information/what-is-genotyping.html#:~:text=Genotyping%20is%20the%20technology%20that,research%2C%20medicine%2C%20and%20
- UKM Medical Molecular Biology Institue (UMBI). (2022). Genetic Variants and Host Risk Factors Associated With Severity of Infection (COVGEN). Kuala Lumpus, WP Kuala Lumpur, Malaysia. Retrieved from https://www.ukm.my/umbi/news/covgen/
- Yin, C. (2020). Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics, 112(5), 3588-3596. doi:https://doi.org/10.1016/j.ygeno.2020.04.016.