
The 2nd AOHUPO Online Educational Series 2022 (Rumusan Laporan)
-
Asia Oceania Human Proteome Organisation (AOHUPO) telah berjaya menganjurkan “The 2nd AOHUPO Online Educational Series 2022” yang diadakan pada tiga hari Jumaat berturut-turut (11, 18, dan 25 Mac 2022) melalui platform webinar. Matlamat utama AOHUPO adalah untuk mempromosi dan menyelaraskan aktiviti komuniti proteomik serantau dan sememangnya ia telah membentuk tradisi penganjuran persidangan dwitahunan yang berjaya oleh negara di rantau Asia Oceania. Pandemik COVID-19 telah menjadi isu kesihatan global dan telah memberi kesan besar kepada pendidikan dan interaksi sosial. Sehubungan itu, pada 2021, Majlis AOHUPO melancarkan program pendidikan atas talian baharu, yang dipanggil “Siri Pendidikan Atas Talian AOHUPO” untuk menyokong pembelajaran dan meneruskan komunikasi semasa pandemik. Tema untuk Siri Pendidikan Atas Talian AOHUPO ke-2 2022 ialah “Renaisans Proteomik Klinikal: Penanda Bio, Pengimejan dan Terapeutik”. Jawatankuasa penganjur yang diketuai oleh pengerusi, Dr. Teck Yew Low (Setiausaha Agung AOHUPO, Universiti Kebangsaan Malaysia) dan pengerusi bersama, Dr Terence Poon (Naib Presiden AOHUPO, Universiti Macau) dan Ho Jeong KWON (Presiden AOHUPO,), telah memberi tumpuan kepada kajian terkini dalam proteomik klinikal. Enam penceramah terkemuka dijemput dalam membentangkan penyelidikan terbaru mereka dalam proteomik klinikal (biomarker, pengimejan dan terapeutik). Berikut adalah laporan mengikut tarikh program (11, 18, dan 25 Mac 2022):
-
Laporan Siri Pendidikan Dalam Talian Pertubuhan Proteom Manusia Asia-Oceania (AOHUPO) ke-2 yang dihoskan di Institut Biologi Molekul Perubatan UKM (UMBI): Rumusan Ceramah untuk Hari yang Pertama (11 Mac 2022)
Ditulis oleh: Profesor Madya Dr. Low Teck Yew
Tarikh penerbitan: 17 May 2022
HARI PERTAMA: 11 MAC 2022
PENGENALAN
Pada tahun 2021, Siri Pendidikan Dalam Talian AOHUPO (AOHUPO OES) telah dilancarkan sebagai acara tahunan dengan matlamat untuk menggalakkan pendidikan saintifik dan kerjasama, pertukaran idea dan budaya, dalam kalangan generasi muda penyelidik proteomik di rantau Asia dan OceaniaPada tahun 2022, jawatankuasa penganjur untuk AOHUPO OES telah memilih “The Renaissance of Clinical Proteomics: Biomarkers, Imaging and Therapeutics” (Kebangkitan Proteomik Klinikal: Penanda Bio, Pengimejan dan Terapeutik) sebagai tema utama untuk AOHUPO OES ke-2. AOHUPO OES ke-2 telah dianjurkan dalam bentuk webinar yang terdiri daripada tiga sesi, di mana setiap sesi berlangsung selama dua setengah jam pada tiga hari Jumaat berturut-turut (11, 18 dan 25 Mac 2022). AOHUPO OES kali ke-2 dihoskan oleh Institut Biologi Molekul Perubatan UKM (UMBI). Enam penceramah antarabangsa terkemuka telah dijemput, meliputi pelbagai aspek proteomik klinikal. Secara keseluruhan, jawatankuasa penganjur menerima pendaftaran daripada 334 peserta yang datang dari 29 negara/wilayah.

Statistik pendaftaran mengikut negara/wilayah geografi untuk Siri Pendidikan Dalam Talian AOHUPO ke-2 2022.
AOHUPO OES dirasmikan bersama oleh Profesor Ho Jeong Kwon, Presiden AOHUPO, dan Profesor Madya Nor Azian Abdul Murad, pengarah UMBI. Sesi ceramah yang pertama telah diberikan oleh Profesor Yoshiya Oda dari Universiti Tokyo, mengenai “Cabaran Pembinaan Hipotesis daripada Multi-Omics: Kes Penyakit Alzheimer” (The Challenge of Hypothesis Building from Multi-Omics: The Case of Alzheimer’s Disease). Profesor Oda bermula dengan memperkenalkan teknik imuno-PCR baru yang dinamakan “Proximity Extension Assay” (PEA). Dalam PEA, protein sasaran diikat oleh dua antibodi khusus; di mana setiap antibodi dikaitkan secara kovalen kepada oligonukleotida yang unik [1]. Selepas pengikatan antibodi-antigen, dua oligonukleotida yang bersebelahan akan berhibrid, dan diikuti oleh PCR kuantitatif. Berbanding dengan LC-MS, PEA telah ditunjukkan sebagai lebih sensitif dalam mengakses bahagian kelimpahan rendah proteom plasma (< 1 ng/ml) [2]. Beliau seterusnya menunjukkan bahawa, daripada 89 sitokin kelimpahan rendah yang disasarkan dalam 350 sampel plasma – 86 sitokin boleh dikesan dalam semua sampel, di mana 79 boleh dikira dalam 50% daripada semua sampel. Selain itu, PEA adalah unggul dari segi daya pemprosesan dan kebolehulangan, di mana CV intra-ujian yang didaftarkan adalah < 10% dalam 90% daripada semua sampel, manakala CV antara ujian adalah < 20% untuk 92% daripada semua sampel. Di samping itu, setiap protein ditentukur untuk julat dinamik linear, dengan had bawah dan atas dicirikan. Menariknya, beliau mendapati tiada pertindihan antara set data LC-MS dan PEA.
Beliau seterusnya membentangkan teknik TMT-lipidomics yang dibangunkan secara dalaman [3], [4]. TMT-lipidomics boleh melabelkan pelbagai kelas lipid yang membawa amina primer, fosfat, dan karbonil sebagai kumpulan berfungsi, dan ia mempunyai daya pemprosesan dan kepekaan yang lebih tinggi sambil mempamerkan keupayaan kuantitatif yang sama seperti kaedah konvensional. Teknik proteomik dan lipidomik ini digunakan untuk menyiasat sampel plasma yang diperoleh daripada subjek warga tua Jepun yang menghidap penyakit Alzheimer (AD) yang berbeza. Analisis data proteomik yang diperolehi dengan LC-MS dan PEA, walaupun tidak menunjukkan pertindihan, telah diperkaya dalam laluan pengaktifan kemokin dan platelet. Ini menunjukkan bahawa perkembangan AD melibatkan pengaktifan platelet oleh kerosakan vaskular, yang seterusnya mendorong pembebasan kemokin. Secara bebas, keputusan ini telah dikolaborasikan oleh data TMT-lipidomics, yang menunjukkan peningkatan regulasi asid hempedu, dan penurunan regulasi DHA/EPA-fosfolipid dan DHA/EPA-trigliserida dalam plasma AD. Ini membawa kepada hipotesis bahawa, dalam AD, penuaan dan kerosakan vaskular mengakibatkan saluran darah bocor pada penghalang usus serta penghalang darah otak. Akibatnya, toksin bocor dari usus ke dalam darah, yang kemudiannya dipindahkan ke penghalang darah otak.

Ceramah yang kedua bertajuk “Menjelajah Proteome yang Belum Dipetakan” (Exploring the Uncharted Proteome) telah dibentangkan oleh Profesor Ruedi Aebersold dari ETH Zurich. Menurutnya, biologi dan perubatan berpusat terutamanya di sekitar fungsi biologi dan fenotip. Dalam era perubatan ketepatan, penjujukan genom telah gagal meramalkan fenotip penyakit daripada gangguan genotip, kerana fenotip ditentukan terutamanya oleh keadaan biokimia dan bukannya genotip. Beliau berpendapat bahawa; protein adalah proksi terdekat kepada fenotip berfungsi. Walau bagaimanapun, penyelidikan proteomik kontemporari tertumpu terutamanya pada mengenal pasti dan mengkuantifikasi protein dan bukannya penyiasatan sistematik dan keseluruhan proteo bagi rangkaian proteoform dan interaksi protein-protein (PPI); walaupun dua yang terakhir lebih dekat dengan fungsi biologi [5]. Tambahan pula, kebanyakan kajian PPI membentangkan model statik interaksi protein, yang tidak mencerminkan perubahan konteks khusus dan dinamik PPI di bawah gangguan yang berbeza. Untuk menyelesaikan masalah ini, makmalnya membangunkan “SEC-SWATH-MS” yang menggabungkan kromatografi pengecualian saiz kepada pemerolehan MS bebas data (DIA) [6]. Dengan teknik ini, protein diekstrak menggunakan keadaan yang sangat ringan untuk mengekalkan PPI. Kompleks protein yang diekstrak kemudiannya tertakluk kepada pemisahan SEC dan pecahan berturut-turut dikumpul dan dianalisis menggunakan SWATH-MS. Untuk menguraikan keputusan, algoritma yang dipanggil SECAT telah dibangunkan untuk menganalisis profil penghijrahan bersama kompleks protein [7]. SECAT berfungsi dengan mengeksploitasi PPI yang diketahui sebagai latar belakang rujukan dan membandingkannya dengan profil elusi SEC-SWATH-MS, untuk menilai kehadiran kompleks yang diketahui dalam gangguan yang berbeza. Menggunakan kaedah ini pada dua varian sel HeLa yang mudah terdedah (CCL4); dan tahan (Kyoto) kepada jangkitan Salmonella Typhimurium, makmalnya mengaitkan rintangan garisan HeLa Kyoto kepada kegagalannya dalam membentuk invadopodium matang akibat kehilangan protein WIPF1 atau WPF2.
Profesor Aebersold seterusnya menerangkan satu lagi algoritma baharu yang dinamakan “Penugasan ProteoForms berasaskan COrelation” (COPF), yang boleh melombong set data DIA bawah ke atas sedia ada untuk mendedahkan bentuk splice alternatif antara gangguan atau keadaan yang berbeza [8]. Ia melakukan ini dengan terlebih dahulu mengukur keamatan peptida merentas banyak sampel; diikuti oleh korelasi berpasangan mereka; pengelompokan dan pemarkahan proteoform. COPF juga boleh digunakan dalam kombinasi dengan SEC-SWATH-MS untuk mendedahkan proteoform khusus keadaan sel yang membentuk kompleks protein yang berbeza.
Rujukan
[1] E. Assarsson et al., “Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability,” PLoS One, vol. 9, no. 4, Apr. 2014, doi: 10.1371/JOURNAL.PONE.0095192.
[2] L. Agrawal, K. B. Engel, S. R. Greytak, and H. M. Moore, “Understanding preanalytical variables and their effects on clinical biomarkers of oncology and immunotherapy,” Semin. Cancer Biol., vol. 52, no. Pt 2, pp. 26–38, Oct. 2018, doi: 10.1016/J.SEMCANCER.2017.12.008.
[3] S. M. Tokuoka, Y. Kita, T. Shimizu, and Y. Oda, “Isobaric mass tagging and triple quadrupole mass spectrometry to determine lipid biomarker candidates for Alzheimer’s disease,” PLoS One, vol. 14, no. 12, Dec. 2019, doi: 10.1371/JOURNAL.PONE.0226073.
[4] S. M. Tokuoka, Y. Kita, M. Sato, T. Shimizu, Y. Yatomi, and Y. Oda, “Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum,” Metabolites, vol. 11, no. 1, pp. 1–15, Jan. 2020, doi: 10.3390/METABO11010019.
[5] I. Bludau and R. Aebersold, “Proteomic and interactomic insights into the molecular basis of cell functional diversity,” Nat. Rev. Mol. Cell Biol., vol. 21, no. 6, pp. 327–340, 2020, doi: 10.1038/s41580-020-0231-2.
[6] I. Bludau et al., “Complex-centric proteome profiling by SEC-SWATH-MS for the parallel detection of hundreds of protein complexes,” Nat. Protoc., vol. 15, no. 8, pp. 2341–2386, 2020, doi: 10.1038/s41596-020-0332-6.
[7] G. Rosenberger et al., “SECAT: Quantifying Protein Complex Dynamics across Cell States by Network-Centric Analysis of SEC-SWATH-MS Profiles,” Cell Syst., vol. 11, no. 6, pp. 589-607.e8, Dec. 2020, doi: 10.1016/J.CELS.2020.11.006.
[8] I. Bludau et al., “Systematic detection of functional proteoform groups from bottom-up proteomic datasets,” Nat. Commun., vol. 12, no. 1, Dec. 2021, doi: 10.1038/S41467-021-24030-X.
-
Laporan Siri Pendidikan Dalam Talian Pertubuhan Proteom Manusia Asia-Oceania (AOHUPO) ke-2 yang dihoskan di Institut Biologi Molekul Perubatan UKM (UMBI): Rumusan Ceramah untuk Hari yang Kedua (18 Mac 2022)
Ditulis oleh: Profesor Madya Dr. Low Teck Yew
Tarikh penerbitan:
HARI KEDUA: 18 MAC 2022
Pada 18 Mac, Dr. Cecilia Lindskog dari Universiti Uppsala membentangkan “Atlas Protein Manusia – proteomik spatial dalam kesihatan dan penyakit” (The Human Protein Atlas – spatial proteomics in health and disease), di mana beliau memperkenalkan Atlas Protein Manusia (www.proteinatlas.org). Dengan 20 tahun pembangunan, akses untuk sumber ini adalah terbuka and percuma. Ia terdiri daripada 10 bahagian, merangkumi (i) tisu, (ii) patologi, (iii) protein darah, (iv) otak, (v) jenis sel tunggal, (vi) jenis sel tisu, (vii) sel imun, (viii) metabolik, (ix) garis sel dan (x) subselular. Beliau memberi tumpuan kepada Atlas Tisu, yang bertujuan untuk mengenal pasti semua protein dalam tubuh badan manusia (10). Atlas Tisu manusia mengandungi pelbagai maklumat seperti imej imunohistokimia mikroarray tisu dalam resolusi sel tunggal, serta data transkriptomi kuantitatif untuk setiap gen pengekodan protein. Pada masa ini, Atlas Tisu merangkumi data protein berasaskan antibodi yang merangkumi 15,323 gen (76%), penyetempatan mereka dalam 78 jenis sel yang berbeza dan 44 jenis tisu; disokong oleh data transkriptomi pukal kuantitatif untuk 37 jenis tisu manusia biasa yang diperolehi secara dalaman (HPA 37 tisu dan 18 jenis sel darah); daripada pangkalan data GTEx (36 tisu) dan FANTOM5 (60 tisu).
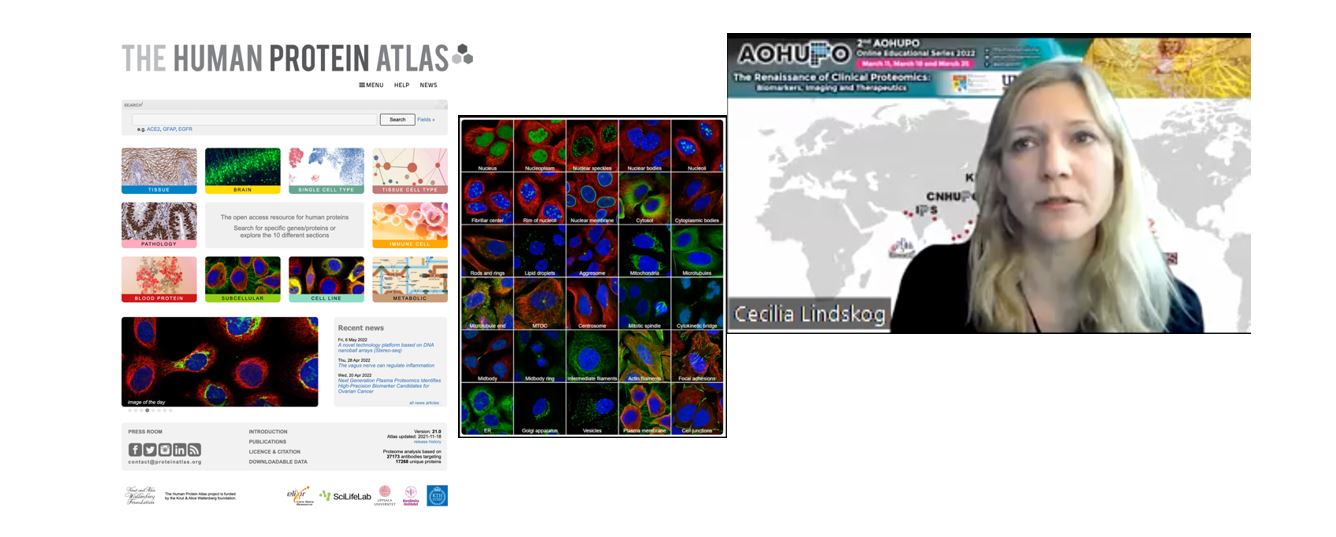
Dr. Cecilia Lindskog menceritakan tentang projek Human Protein Atlas yang telah dibangunkan oleh pasukan di Sweden selama 20 tahun.
Untuk menentukan semua gen manusia dalam sesuatu tisu yang terpilih, tahap ekspresi dan pengedaran spatial mRNA dibahagikan kepada 5 kategori pengayaan (tisu diperkaya; kumpulan diperkaya; tisu dipertingkatkan, kekhususan rendah dan tidak dikesan), boleh dilihat sebagai carta bar. Baru-baru ini, pengelompokan gen UMAP juga disediakan untuk melihat corak ekspresi jiran gen terdekat setiap jenis tisu. Versi terkini (Ver. 21) Atlas Tisu juga menggabungkan data transkriptomi sel tunggal (sc-RNA-seq). Ini disebabkan organ/tisu sendiri terdiri daripada banyak jenis sel, oleh itu data transkriptomi pukal sahaja tidak menawarkan resolusi yang mencukupi. Sehingga kini, data sc-RNA-seq meliputi 25 tisu manusia dan sel darah, termasuk 444 kelompok jenis sel tunggal dan 76 jenis sel utama. Baru-baru ini, makmal Dr. Lindskog menggunakan sumber ini untuk mengkaji enzim penukar angiotensin I manusia 2 (Angiotensin Converting Enzyme 2; ACE2), reseptor permukaan sel yang dicadangkan sebagai terlibat dalam kemasukan sel hos untuk virus SARS-CoV2 [1]. Transkriptomi pukal menunjukkan bahawa ACE2 dinyatakan dengan banyak dalam usus kecil, kolon dan duodenum tetapi tidak di dalam paru-paru. Data ini disokong oleh data sc-RNA-seq yang diperolehi daripada pelbagai jenis sel konstituen. Pemerhatian ini telah disahkan oleh imunohistokimia yang dilakukan dengan 2 antibodi berbeza yang menyasarkan ACE2, daripada 45 jenis tisu normal dan 159 jenis sel berbeza daripada ~ 800 individu.
Seterusnya, Profesor Kathryn Lilley dari Universiti Cambridge membentangkan “Organisasi Spatial Sel “ (The Spatial Organization of the Cell). Protein dan RNA mesti bergerak ke lokaliti selular yang betul untuk melaksanakan fungsinya yang sepatutnya; dan penyetempatan protein/RNA yang menyimpang boleh berakhir dengan penyakit. Ciri-ciri yang mempengaruhi penyetempatan protein termasuk urutan asid amino; pengubahsuaian selepas transkripsi dan pasca terjemahan; interaksi dengan biomolekul lain, lokasi di mana protein diterjemahkan dan konformasi protein. Oleh itu, perubahan dalam ciri-ciri ini, serta gangguan alam sekitar sering mengakibatkan penyetempatan protein alternatif/menyimpang. Makmal Profesor Lilley berminat dengan cara protein/RNA berakhir di alamat yang betul; bagaimana ciri biomolekul mempengaruhi lokaliti mereka, dan sama ada protein/RNA menjalankan fungsi yang sama di lokasi yang berbeza. Makmal beliau telah membangunkan kaedah dan algoritma untuk mencirikan penyetempatan protein dan RNA secara seluruh sistem.
Untuk mengenal pasti lokaliti subselular proteom, kaedah yang dipanggil LOPIT (Localization of Protein using Isotope Tagging) digunakan. LOPIT bergantung pada pecahan subselular berasaskan ultrasentrifugasi bagi komponen selular, diikuti dengan pelabelan isotop bagi setiap pecahan trypsin dan pengenalpastian dan kuantifikasi berasaskan LC-MS. Selepas itu, profil korelasi protein antara pecahan berturut-turut ditentukan, di mana protein yang mempamerkan profil serupa dianggap sebagai berasal dari organel yang sama [2]. Secara teknikal, pecahan subselular boleh dicapai dengan ultrasentrifugasi kecerunan ketumpatan keseimbangan (HyperLOPIT), yang memerlukan sekurang-kurangnya 200 juta sel walaupun menawarkan resolusi subselular yang unggul; atau ultrasentrifugasi pembezaan (LOPIT-DC), yang memerlukan kurang bahan permulaan (50-70 juta sel) [3], [4]. Dari segi fungsi, LOPIT boleh digunakan untuk menjana “peta statik” atau “peta dinamik” untuk proteom yang di bawah kajian. Walaupun peta statik biasanya digunakan untuk mengenal pasti protein kepada lokaliti subselular di bawah keadaan tertentu, atau untuk mengenal pasti organel baharu atau kompleks subselular yang kurang dicirikan, peta dinamik digunakan untuk menyiasat penyetempatan semula khusus protein atas gangguan selular, penyakit, atau peringkat perkembangan. Dalam kajian baru-baru ini, monosit THP-1 telah dirawat dengan lipopolisakarida (LPS) untuk mendorong tindak balas keradangan, diikuti oleh HyperLOPIT untuk menghasilkan peta dinamik [5]. Menariknya, antara 253 protein yang disetempatkan semula, hanya 2 perubahan yang didaftarkan dalam kelimpahan, membayangkan bahawa mengukur kelimpahan protein sahaja boleh disalahkan. Akhirnya, kedua-dua protokol LOPIT telah diubahsuaikan untuk menyiasat penyetempatan (semula) transkrip, dan menghasilkan kaedah menimbulkan LoRNA (Localization of RNAs), yang bertujuan untuk menetapkan transkrip termasuk kedua-dua RNA pengekodan dan bukan pengekodan kepada semua lokaliti subselular [6]. Kedua-dua LOPIT dan LoRNA boleh dilakukan dalam satu langkah untuk menyetempatkan protein dan RNA secara serentak.
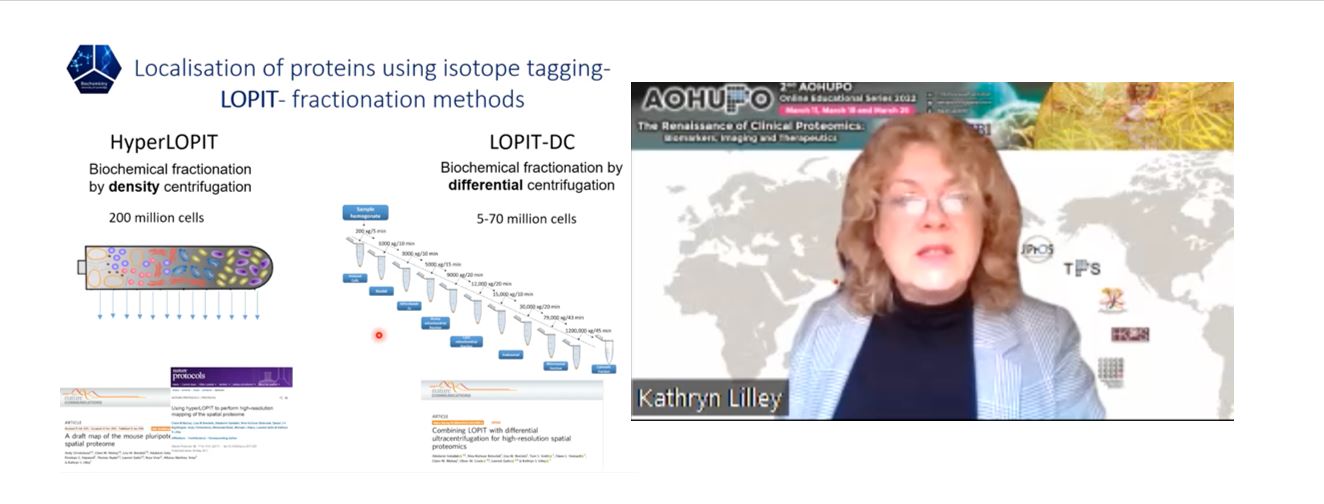
Profesor Kathryn Lilley dari Cambridge. Beliau adalah perintis yang mencipta kaedah mencirikan penyetempatan proteom yang dinamakan LOPIT.
Rujukan
[1] F. Hikmet, L. Méar, Å. Edvinsson, P. Micke, M. Uhlén, and C. Lindskog, “The protein expression profile of ACE2 in human tissues,” Mol. Syst. Biol., vol. 16, no. 7, Jul. 2020, doi: 10.15252/MSB.20209610.
[2] T. P. J. Dunkley, R. Watson, J. L. Griffin, P. Dupree, and K. S. Lilley, “Localization of organelle proteins by isotope tagging (LOPIT),” Mol. Cell. Proteomics, vol. 3, no. 11, pp. 1128–1134, Nov. 2004, doi: 10.1074/mcp.T400009-MCP200.
[3] A. Christoforou et al., “A draft map of the mouse pluripotent stem cell spatial proteome,” Nat. Commun., vol. 7, Jan. 2016, doi: 10.1038/NCOMMS9992.
[4] A. Geladaki et al., “Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics,” Nat. Commun., vol. 10, no. 1, Dec. 2019, doi: 10.1038/S41467-018-08191-W.
[5] C. M. Mulvey et al., “Spatiotemporal proteomic profiling of the pro-inflammatory response to lipopolysaccharide in the THP-1 human leukaemia cell line,” Nat. Commun., vol. 12, no. 1, Dec. 2021, doi: 10.1038/S41467-021-26000-9.
[6] E. Villanueva et al., “A system-wide quantitative map of RNA and protein subcellular localisation dynamics,” bioRxiv, p. 2022.01.24.477541, Jan. 2022, doi: 10.1101/2022.01.24.477541.
-
Laporan Siri Pendidikan Dalam Talian Pertubuhan Proteom Manusia Asia-Oceania (AOHUPO) ke-2 yang dihoskan di Institut Biologi Molekul Perubatan UKM (UMBI): Rumusan Ceramah untuk Hari yang Ketiga (25 Mac 2022)
Ditulis oleh: Profesor Madya Dr. Low Teck Yew
Tarikh penerbitan:
HARI KETIGA: 25 MAC 2022
Dalam ceramah beliau yang bertajuk “Menerokai Interaksi Fenotip-Sasaran Ubat dengan Pendekatan Proteomik berasaskan MS dan Kesan Translasinya” (Exploring Drug-Target-Phenotype Interaction with MS-based Proteomics Approaches and its Translational Impacts), Profesor Ho Jeong Kwon dari Universiti Yonsei menerangkan bagaimana beliau memilih molekul kecil sebagai alat untuk menerokai proteom. Alam semulajadi membekalkan kita banyak jentera biosintetik yang menghasilkan molekul kecil yang boleh berkomunikasi dengan biomolekul asing seperti protein untuk menimbulkan kesan biokimianya, satu proses yang dinamakan “Sosiologi Molekul”. Interaksi sedemikian boleh dieksploitasi untuk aplikasi terapeutik, dan dia memetik Taxol, ubat kemoterapi yang diperoleh daripada sumber semula jadi sebagai contoh. Strategi utama untuk penemuan ubat di makmal beliau melibatkan langkah-langkah yang berikut: (i) skrin fenotip berasaskan sel dengan perpustakaan kimia untuk mengenal pasti molekul kecil sasaran yang boleh mengganggu fenotip terpilih; (ii) proteomik kimia untuk mengenal pasti sasaran protein yang mengikat molekul kecil sasaran ini; (iii) pengesahan pengikatan dadah-protein dengan bioinformatik struktur; dan aktiviti dan ujian fungsian; (iv) mereka-bentuk ubat yang lebih baik dan mewujudkan platform untuk mempercepatkan penemuan ubat; dengan itu memberikan (v) maklum balas untuk menemui ubat baharu yang menyasarkan proses biologi baharu.
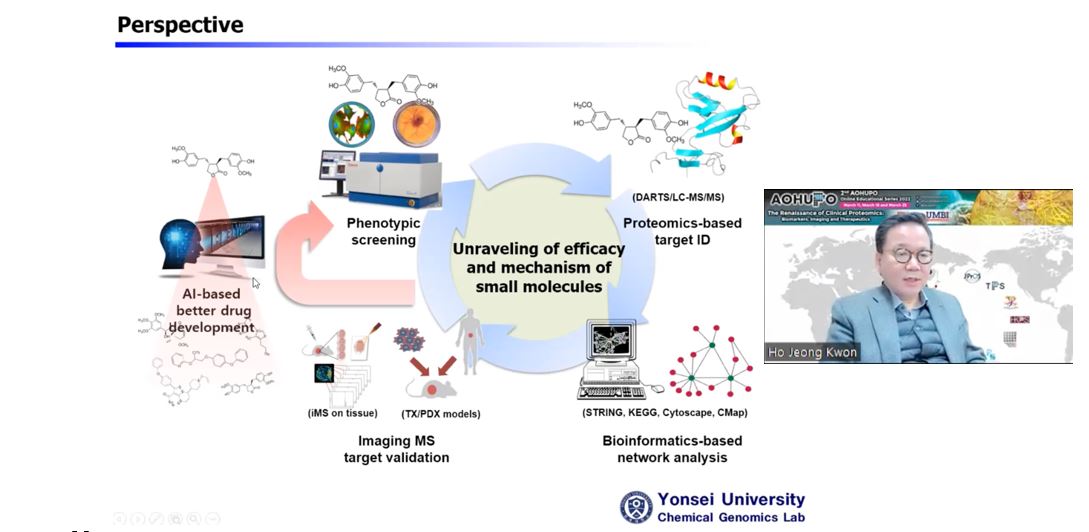
Profesor Ho Jeong Kwon dari Universiti Yonsei membentangkan ucaptama beliau. Makmal beliau mengeksploitasikan molekul kecil untuk menyiasat fungsi proteom.
Beliau menghuraikan prosedur untuk menemui molekul kecil yang menyasarkan autophagy, proses degradasi diri yang berkaitan dengan kanser dan penyakit neurodegeneratif. Dalam proses ini, makmal beliau pertama kali melakukan skrin fenotip dengan perpustakaan kimia terhadap sel HeLa, diikuti oleh pewarnaan Acridine Orange (AO) yang mengesan aktiviti lisosom [1]. Ini mengakibatkan penemuan beberapa induser autophagy termasuk Kaem, Sert, Rg3 dan CTS. Untuk mengenal pasti protein sasaran molekul kecil ini, makmalnya menggunakan kaedah proteomik kimia bebas pengubahsuaian seperti DARTS (Drug Affinity Responsive Target Stability) dan CETSA (Cellular Thermal Shift Assay) [2]. Kedua-dua kaedah adalah berdasarkan premis bahawa pengikatan molekul kecil memberikan perubahan konformasi, serta kestabilan protease dan haba kepada protein, menjadikannya kurang terdedah kepada proteolisis (DARTS) dan denaturasi haba (CETSA) berbanding populasi yang tidak terikat. Pembacaan untuk perubahan ini boleh didaftarkan dengan membandingkan proteom yang dirawat dengan ubat dan tidak dirawat menggunakan proteomik berasaskan MS kuantitatif atau western blot. Akhirnya, beliau menamatkan ceramah dengan menerangkan bagaimana pengimejan MS boleh digunakan untuk menyetempatkan pengedaran pengedaran molekul kecil dalam tisu hidup, dan bagaimana tarikh ini boleh dianalisis secara kolektif oleh AI untuk mereka bentuk ubat yang lebih baik [3].
Dalam ceramah yang terakhir – “Pengenalpastian ligan HLA melalui Imunopeptidomik berasaskan MS untuk Pembangunan Imunoterapi Kanser Peribadi” (Identification of HLA ligands through MS-based Immunopeptidomics for Development of Personalized Cancer Immunotherapy), Dr. Michal Bassani-Sternberg dari Institut Penyelidikan Kanser Ludwig di Lausanne memperkenalkan penemuan antigen tumor, dan bagaimana ia boleh membantu membangunkan Terapi pemindahan sel T dan vaksin kanser. Beliau menggunakan imunopeptidomik berasaskan MS kepada sampel tumor untuk membongkar peptida terikat HLA-I dan HLA-II yang dinamakan antigen berkaitan tumor (TAA), yang agak terhad tetapi tidak unik kepada sel tumor (20). HLA adalah poligenik dan polimorfik, dengan setiap bentuk mengikat peptida antigen yang membawa motif jujukan yang ditentukan, yang kini boleh diramalkan.

Dr. Michal Bassani-Sternberg menceritakan aliran kerja immunopeptidomik yang diamalkan di makmal beliau untuk mengenal pasti antigen spesifik tumor yang ada pada pesakit kanser.
Makmal beliau telah mewujudkan aliran kerja yang memurnikan imun secara berurutan, kedua-dua peptida HLA-I dan HLA-II untuk pemerolehan MS [4]. Untuk melanjutkan aliran kerja ini kepada pengesanan neoantigen khusus tumor, makmalnya turut menggabungkan urutan DNA dan RNA tisu yang sama untuk membina pangkalan data proteogenomik tersuai yang dilampirkan dengan varian jujukan daripada sampel individu. Selepas itu, imunogenisiti selular epitop yang dikesan disahkan, dan vaksin kanser dan imunoterapi peribadi pemindahan sel T boleh dibangunkan. Keputusan yang diperolehi daripada saluran paip proteogenomik ini membolehkannya mengenal pasti antigen berkaitan tumor (TAA) dan antigen spesifik tumor (TSA) yang boleh diambil tindakan; serta untuk meningkatkan prestasi algoritma ramalan HLA dalam silico [5], [6]. Baru-baru ini, makmal beliau turut mengembangkan aliran kerja proteogenomik untuk menggabungkan jujukan bukan kanonik, yang pada asalnya dianggap sebagai tanpa pengekodan protein, contohnya, RNA bukan pengekodan, kawasan 3’ dan 5’ yang tidak diterjemahkan dan unsur transposon. Saluran paip baharu ini menggunakan penjujukan exome, RNA-seq dan RIBO-seq; dan pangkalan data diperibadikan yang terhasil mengandungi jujukan daripada terjemahan tiga bingkai RNA [7]. Untuk menangani saiz pangkalan data yang melambung dan isu FDR, makmal beliau membangunkan enjin carian baharu yang dipanggil NewAnce yang menggabungkan MaxQuant dan COMET. Selain itu, data RIBO-seq juga digunakan untuk mengesan penterjemahan sebenar supaya pangkalan data tersuai yang lebih kecil dan lebih tepat boleh dibina untuk mengurangkan FDR.
Rujukan
[1] D. Kim, H. Y. Hwang, E. S. Ji, J. Y. Kim, J. S. Yoo, and H. J. Kwon, “Activation of mitochondrial TUFM ameliorates metabolic dysregulation through coordinating autophagy induction,” Commun. Biol., vol. 4, no. 1, Dec. 2021, doi: 10.1038/S42003-020-01566-0.
[2] J. Chang, Y. Kim, and H. J. Kwon, “Advances in identification and validation of protein targets of natural products without chemical modification,” Nat. Prod. Rep., vol. 33, no. 5, pp. 719–730, May 2016, doi: 10.1039/C5NP00107B.
[3] T. Y. Kim et al., “Matrix-assisted laser desorption ionization – mass spectrometry imaging of erlotinib reveals a limited tumor tissue distribution in a non-small-cell lung cancer mouse xenograft model,” Clin. Transl. Med., vol. 11, no. 7, Jul. 2021, doi: 10.1002/CTM2.481.
[4] H. S. Pak et al., “Sensitive Immunopeptidomics by Leveraging Available Large-Scale Multi-HLA Spectral Libraries, Data-Independent Acquisition, and MS/MS Prediction,” Mol. Cell. Proteomics, vol. 20, 2021, doi: 10.1016/J.MCPRO.2021.100080.
[5] M. Bassani-Sternberg et al., “Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry,” Nat. Commun., vol. 7, Nov. 2016, doi: 10.1038/NCOMMS13404.
[6] M. Bassani-Sternberg et al., “Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity,” PLoS Comput. Biol., vol. 13, no. 8, p. e1005725, Aug. 2017, doi: 10.1371/JOURNAL.PCBI.1005725.
[7] C. Chong et al., “Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes,” Nat. Commun., vol. 11, no. 1, Dec. 2020, doi: 10.1038/S41467-020-14968-9.